This article had been covered by IP Fray Law360 and World IP Review (WIPR).
On 21 November 2023, Mathys & Squire lodged a request under Rule 262.1(b) of the UPC’s Rules of Procedure requesting that the Court make available all written pleadings and evidence filed in relation to a case between Astellas and Healios KK. Finally, on 4 November 2024, over 11 months later, the UPC court have ordered that Mathys & Squire should have access to the pleadings as requested in unredacted form.
Public access to pleadings and evidence filed with the UPC registry is written into the Unified Patent Court Agreement with Article 45 UPCA promising that proceedings “shall be open to the public unless the Court decides to make them confidential, to the extent necessary, in the interest of one of the parties or other affected persons, or in the general interest of justice or public order.”
The extensive delays in granting Mathys & Squire access to court documents demonstrate the extent to which this promised public access in anything approaching a reasonable timescale has proven illusory in relation to ongoing cases as the following timeline of the case demonstrates.
| 21 Nov 2023 | Mathys & Squire files public access request |
| 12 Dec 2023 | Court issues preliminary order informing parties of an intention to suspend the application pending a decision in Ocado v Autostore appeal |
| 28 Dec 2023 | Case suspended pending a decision in Ocado v Autostore |
| 10 Apr 2024 | Ocado v Autostore decision issues – case restarts |
| 1 May 2024 | Mathys & Squire provide Court with comments on Ocado v Autostore |
| 22 May 2024 | Court sets 5 June deadline for parties’ responses |
| 5 Jun 2024 | Astellas provides comments on Ocado v Autostore appeal |
| 19 Jun 2024 | Astellas and Healios KK inform Court of out of court settlement |
| 24 Jul 2024 | Court publishes an order confirming settlement of underlying proceedings and sets date of 16 August for parties to comment on access request if Mathys & Squire confirm that the access request is maintained |
| 22 Aug 2024 | Court orders Mathys & Squire to be provided access to the court documents subject to redactions requested by Astellas and additional redactions imposed by Court |
| 12 Sept 2024 | Copies of redacted documents provided to Mathys & Squire |
| 1 Oct 2024 | Mathys & Squire files a request to have Astellas’ redactions removed |
| 3 Oct 2024 | Mathys & Squire files request for Court to provide copies of unpublished orders referred to in the case |
| 17 Oct 2024 | Mathys & Squire files request for Court to remove erroneous Court-imposed redaction and provide copies of erroneously omitted documents |
| 4 Nov 2024 | Court orders Astellas’ redactions removed, provides missing documents and removes erroneous Court redactions |
When access to the pleadings was initially granted in September, the documents were subjected both to redactions that had been requested by Astellas’ representatives and those imposed by the Court.
Review of the documents revealed that some documents filed during the case had not been provided to us. Indeed, those documents were not listed in the electronic case management system and their existence only became apparent because they were referred to elsewhere in the pleadings. Review of the documents also revealed that many of the redactions imposed by the court were inconsistent, unnecessary and clearly went beyond those necessary in the interests of any of the parties or the general interest of justice or public order.
Examples of some of the more bizarre redactions imposed by the court included redactions of names of commercially-available laboratory equipment and reagents, standard experimental protocols, arbitrary portions of headers, figure labels and tables of contents. Titles of academic papers cited as prior art and identified by the names of their authors had also been randomly redacted, as had routine publicly-available information such as the names of inventors and public officials, and random portions of electronic signature blocks. In many cases redaction had been inconsistent, with the same information being redacted or not redacted in different copies of documents provided by the Court.
Examples of this highly inconsistent approach to redaction are illustrated below.
A). Inventors’ names redacted from translation of priority document while left unredacted in Japanese original in copy provided by Court


B). Two versions of a signature block included in a document provided by the Court multiple times – one with redactions, a second where the Court considered that redactions were not necessary


C). Examples of Court redactions of titles of prior art documents


D). Random redaction of names of laboratory equipment manufacturers from documents provided by Court


E). Partial redaction of figure labels in a publicly-available prior art document
It also appeared from the redactions that the Court intended to keep the identities of the parties’ expert witnesses confidential; however, this objective was defeated by the fact that the witnesses’ names were visible in the filenames given to their witness statements as listed in the case management system.
In addition, reading through the documentation, it was apparent that the pleadings and evidence referred to multiple Court orders which, contrary to the UPC Rules of Procedure, had not been published.
Further correspondence with the Registrar has revealed the causes of some, but not all, of these errors.
Apparently, the Court uses an anonymization tool which should theoretically only redact personal information present in pleadings and evidence. However, as is evidenced by the above, this tool is unreliable and information is misidentified leading to errors. Only by making a further application to the Court could (some of) these erroneous redactions be removed.
The Court’s failure to publish court orders as required by Rule 262.1(a) of the Court’s Rules of Procedure, which states that “decisions and orders made by the Court shall be published”, arises due to a conflict between this rule and Rule 34.2 (c) of the Rules Governing the Registry of the Unified Patent Court. That rule imposes on the Registrar an obligation to ensure that the UPC website contains a “collection of the final decisions and orders of the Court of general interest, as well as any corresponding translations of the headnotes and/or decisions in English, French and German”. This has been interpreted by the Court as limiting the decisions and orders which they need to publish. The hierarchy between the Rules of Procedure and the Rules Governing the Registry is not clear, but the Registry obviously considers itself to be bound primarily by the latter.
The practical effect of the Registry’s interpretation of the Rules is that it is incredibly difficult for third parties to monitor the progress of cases through the Court. Most of the orders the Court issues are orders specific to a case which would inform third parties regarding how a case is proceeding. But none of those orders ever see the light of day.
Regrettably, the Court’s Rules of Procedure place significant barriers on any third parties wanting to establish what is happening in a case before the Court. In February 2024, the Court interpreted its rules as requiring third parties wanting access to court documents to go to the expense of engaging representatives in order to file such requests. Since then, several divisions of the Court have taken (e.g. here, here, and here) a narrow view of the public interest in obtaining information about ongoing court cases and an expansive view of excluding the public from access to pleadings. These decisions are based on the grounds of the potential impact on the integrity of proceedings should members of the public actually obtain access to documents filed with the Court.
The Court took a very leisurely approach to progressing our access request, taking nearly a year to provide the requested documents. The result of the delays was that the case we wished to observe was settled before we obtained sight of any pleadings.
The Court’s erroneous redactions and the Registrar’s failure to comply with the Court’s orders then required us to make yet further applications to see the pleadings in full, as did a request by one of the parties to redact a portion of the pleadings, a request which the Court ultimately found to be unjustified.
Ultimately, the access request was successful. However, the extensive submissions required to obtain sight of the pleadings demonstrates the difficulties that third parties have in obtaining any information about disputes pending before the Court.
A recent report by the European Patent Office [1] has revealed that 10% of all inventions for which a European patent application has been filed are generated by university research conducted in Europe (measured in terms of number of patent applications filed at the European Patent Office).
The report is based on data collected between 2000 and 2020 featuring 1,200 European universities that have directly filed patent applications at the EPO, or that have university-affiliated researchers named as inventors on ‘indirect’ patent applications. Roughly two-thirds of the ‘university-generated’ patent applications fall into this ‘indirect’ category – i.e., the applications are filed by other entities such as small and medium-sized enterprises (including spin-out companies), not by the universities themselves.
The UK seems to perform consistently well, and ranks third for the total number of universities filing European patent applications per country, and number of academic European patent applications filed per country overall (following Germany and France). Of the 131 UK universities that filed at least one European patent application between 2000-2020, the University of Oxford is the UK’s leading institution, filing the most European patent applications of any UK-based university (1,660 in total). The average number of European patent applications filed by a UK-based university is approximately 100, but the data is heavily skewed: four universities filed over 1,000 patent applications each, and only six others filed between 300-1000 patent applications each. Of course, there will be differences in size and resources between universities, but some institutions may be missing opportunities to exploit and monetize university-generated IP more widely.
Statistics on applications made jointly by universities with a co-owner may shed further light on this skew in the UK data: in France, 79% of university-originating patent applications are filed with a co-owner, frequently a large public research organisation such as CNRS or INSERM. In contrast, only 10% of British universities file patent applications with a co-owner. Given the similarities between the sizes of the British and French populations and economies, one interpretation of this difference might suggest that UK university IP currently being left by the wayside could be unlocked by an enhanced role for UK public research organisations, such as the research councils or UKRI, in the commercialisation of research. Alternatively, smaller UK universities with limited resources could create shared technology transfer organisations to fulfil a similar role, as recommended in the UK Government’s recent independent review of university spin-outs. [2]
It is encouraging to see that UK startup businesses are particularly well-represented in the study. For example, 281 UK startups filed a total of 853 European patent applications relating to university-generated inventions between 2015 and 2019 (more startups and more startup-filed applications than any other country in the study).
In addition, four UK universities appear in the top eight academic institutions associated with startups that have filed patent applications to university-generated inventions: Oxford is joined by the University of Cambridge, Imperial College London, and University College London (perhaps unsurprisingly, the same four universities who filed more than 1000 European patent applications each across the study period). Indeed, the University of Cambridge is the second highest in this particular list (associated with 93 startups who filed European patent applications between 2000-2020) – just pipped to the post by ETH Zurich (101 startups).
Despite this success, the report comments on the “European paradox” and the difficulties of transforming advantages in academic research into applied technological and economic performance, compared to other advanced economies such as the US. The report suggests one reason for this is the difference in startup landscape between the US and Europe. The study also references Mario Draghi’s report on the future of European competitiveness [3] and Enrico Letta’s report on the future of the Single Market [4], which point to a fragmented innovation ecosystem across Europe as being central to Europe’s struggle to translate innovation into commercialisation. That 10% of startups with European academic patents are headquartered in the US (as reported in the EPO’s findings) highlights this struggle, as does Imperial College’s launch of a new science and tech hub in San Francisco [5]. Further emphasising the apparent ease of startup building and scaling in the US compared to Europe is Entrepreneur First (EF)’s recent development – whilst first launched in Europe in 2011, EF now requires all its startups to move to California for part of the EF programme and to officially incorporate their companies in the US. Changes to the innovation ecosystem are needed to retain startups using university-generated innovations in Europe.
To try and aid connections between potential investors and startups, the EPO Observatory on Patents and Technology has launched the Deep Tech Finder (DTF) [6], a digital platform designed to easily identify and analyse startups based within EPO member states that have filed European patent applications (and who may be seeking funding or business partners).
At Mathys & Squire, we understand that startup and scaleup businesses are at the frontline of innovation. We have expertise in supporting such businesses identify, protect and commercialise IP in every industry and sector. If you would like to find out more about how we can help you with your IP needs, please get in contact with our dedicated ‘Scaleup Quarter’.
[1] “The role of European universities in patenting and innovation”
[2] Independent review of university spin-out companies – GOV.UK
[3] EU competitiveness: Looking ahead
[4] Much more than a market report by Enrico Letta
[5] Imperial to strengthen Transatlantic tech cooperation with new hub
[6] Explore deep tech in Europe – EPO
About a year ago, the Enlarged Board of Appeal issued its decision in G 1/22 (consolidated with G 2/22), which significantly relaxed the EPO’s approach to ‘same applicant’ priority. Under the old approach, life could be tough: a priority claim was deemed invalid if a proprietor was unable to show, when challenged, that the applicants for the subsequent application included all of the applicants for the priority application or their successor(s) in title at the time the subsequent application was filed. Then the sun came out: the Enlarged Board held that there is a strong, rebuttable presumption that the priority applicants approve of the subsequent applicants’ entitlement to priority, regardless of any difference in names. See our earlier report here.
The CRISPR cases provide a striking example of this shift in the law.
Prior to G 1/22, Board of Appeal 3.3.8 upheld a decision of the EPO opposition division revoking an important CRISPR patent belonging to Broad, MIT and Harvard on the basis of intervening art that only became citeable because the subsequent applicants did not include one of the priority applicants or its successor title (and notwithstanding a plea from the proprietors for the EPO to take a more relaxed approach to ‘same applicant’ priority). See our reports of T 844/18 here and here.
Now, post-G 1/22, the same issue has come before Board 3.3.8 (albeit in a slightly different composition) in the context of a divisional of the revoked patent in case T 2360/19 (consolidated with T 2516/19 and T 2689/19, where the same issue also arose). How did the proprietors fare? Much better. The Board held that the omission of the relevant party from the subsequent application was of no consequence in view of the strong presumption in favour of priority entitlement. Essentially the same EPO tribunal with essentially the same set of facts before it came to the opposite conclusion on priority.
T 2360/19 is also noteworthy because of the Board’s finding that evidence of a dispute between the proprietors and the omitted party was not sufficient to rebut the strong presumption in favour of priority.
The dispute arose because the proprietors had filed the subsequent application (the PCT application from which the European family derived) without naming one of the priority applicants (Luciano Marraffini) or his successor in title (the Rockefeller University) on the PCT request form. The dispute was settled in 2018 by an arbitrator who decided that Marraffini should not be named as an inventor and Rockefeller should not be named as a proprietor of the PCT application. However, the opponents argued that the existence of the dispute proved there was no agreement on the transfer of priority rights belonging to Marraffini/Rockefeller.
The Board was of the contrary view – it saw the dispute as supporting the presumption of validity of the priority claim, because Marraffini and Rockefeller had wanted to be named in the PCT application, and therefore it was not credible that either of them would have acted in a way to invalidate the priority claim, particularly when the very purpose of the arbitration settlement was to safeguard the inventions made by Marraffini and others. The Board furthermore identified the settlement as an ex-post (retroactive) agreement on the transfer of priority rights, in line with the Enlarged Board’s suggestion in G 1/22 that the EPO should accept such agreements.
Interestingly, the settlement was not actually decisive to the outcome in T 2360/19 though. The Board held that the result would have been the same even in the absence of the settlement evidence, because “As also reiterated in G 1/22 … There is always a party who is entitled to claim priority, even if this party has to be determined in a national proceedings (with this being the same if the dispute is settled outside the courts, by way of amicable settlement or arbitration, as is the case here) … only the rebuttable presumption of a priority right guarantees that … this right is not ‘lost’ somewhere in an inventorship dispute.” The logic seems to be that, to the extent such a dispute has any bearing on the claim to priority of a patent, then the presumption is that the outcome of the dispute will entail preservation of the priority claim. This chimes with the Enlarged Board’s comment in G 1/22 that the EPC explicitly foresees the ex tunc assignment of priority rights in the context of disputes on the right to the patent before national courts, and that, in view of legal certainty, third parties can never fully rely on the invalidity of a priority claim.
What would it take to rebut the presumption that the subsequent applicants are entitled to claim priority? The Enlarged Board gave the example of party acting in bad faith. Thus, if evidence is provided that subsequent applicants filed a subsequent application without the agreement of a priority applicant or its successor in title, and that the priority applicant/successor does not and never did agree to the priority claim, then this might be enough to rebut the presumption. However, that is very different from the situation in T 2360/19, where the proprietors filed a statement from Rockefeller after the arbitration was concluded confirming that the university had been aware that the other proprietors were filing a series of PCT applications on CRISPR technology based on an inventorship determination, and that the university agrees that the correct parties were named on those PCT applications.
The case in T 2360/19 has now been remitted to the opposition division for examination of the other grounds of opposition. The decision, and that in each of the other consolidated cases, is likely to have broader significance for the proprietors though, because they omitted Marraffini/Rockefeller from many other PCT applications that contain the same priority claims. Whether the proprietors fare well before the opposition division following remittal remains to be seen – Board 3.3.8 recently issued a preliminary opinion in which it found a lack of enabling disclosure in the priority application for a similar CRISPR patent belonging to the University of California and others, which the proprietors then self-revoked, as reported here.
Commentary by Partner Edd Cavanna has been featured in Law360, World Intellectual Property Review, The Register and Communications Today discussing the global increase of semiconductor patent applications following the growth of the AI sector.
Read the extended press release below.
Background
There has been a 22% increase in global semiconductor patents filed, rising from 66,416 in 2022/3* to 80,892 in 2023/4 (year-end March 31), shows new research by Mathys & Squire, the leading intellectual property law firm.
Dr Edd Cavanna, partner at Mathys & Squire, says the rise in new inventions in the sector is partly being driven by the boom in the AI sector which has dramatically increased the market cap of chip companies such as Taiwan Semiconductor Manufacturing.
Dr Cavanna says: “Gen-AI is the most recent technology that is spurring R&D in the semiconductor industry and leading to an associated rise in patent applications.”
Chinese patents up 42% as domestic industry responds to US export restrictions of some semiconductors
Global semiconductor patent applications filed in China have risen 42% to 46,591 in 2023/4, up from 32,840 the year before.
The sharp uptick in Chinese applications is in keeping with a general trend recently of strong growth and large filing numbers. In contrast, new US patent application filings have remained reasonably steady over the past few years.
Dr Cavanna says: “This is likely an indication that the rivalry between US and China in the semiconductor patent space is heating up.”
Export restrictions on some semiconductors from the US to China and concerns that more export restrictions could be announced in the future may have encouraged more investment and R&D from the Chinese semiconductor industry.
The Chinese government has previously called for China to ‘step up’ progress in the innovation of key technologies, including semiconductors as well as AI research which depends heavily on semiconductor technology.
Dr Cavanna says: “The massive rise in semiconductor patent applications in China also shows a vote of confidence amongst the semiconductor industry in China as a market for semiconductors.”
In the US the Inflation Reduction Act (IRA) may also have helped trigger more R&D in the sector. The IRA provides substantial financial incentives, running into billions of dollars in subsidies, to chip manufacturers to manufacture more semiconductors in the US. That funding may be helping more R&D in the sector. Semiconductor patents in the US have risen 9% to 21,269 in 2023/4.
Export restrictions have also led to an increase in chip manufacturing from foreign firms in the US. For example Taiwan Semiconductor Manufacturing Co, one of the largest manufacturer of chips, has undertaken an unprecedented investment in constructing an Arizona plant that reportedly rivals the productivity of its plants in Taiwan.
There has been a 22% rise in patents applications for semiconductors from 2022/3 to 2023/4
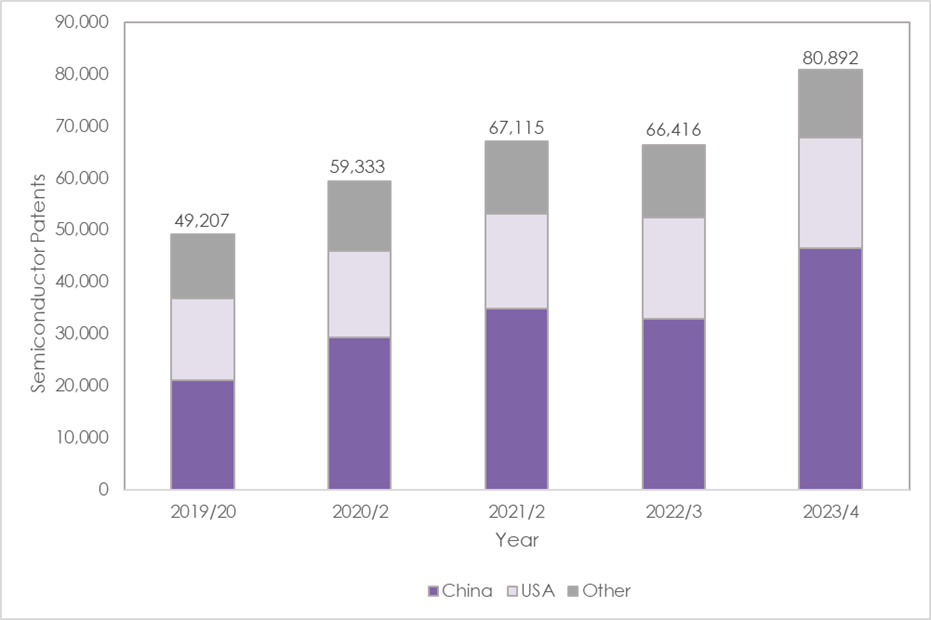
* Applications filed in the year ending March 31, Source: World Intellectual Property Organization
Historically, women’s health has been greatly underfunded and under-researched. Nowhere is this gender-biased neglect more evident than in the research and clinical management of menopause symptoms. In response, World Menopause Day, held every year on 18 October, aims to raise awareness of the symptoms and impact of menopause and highlight the options available for supporting those going through the menopause transition.
The World Health Organisation (WHO) defines menopause as “the permanent cessation of menstruation resulting from loss of ovarian follicular activity”.
In the UK, the average age for menopause is 51. However, spontaneous (natural) early menopause affects approximately 5% of the population before the age of 45 whilst premature menopause, defined as menopause before the age of 40, occurs in approximately 1% of women. It has been estimated that over one billion women globally will be perimenopausal or postmenopausal by 2030 and nearly 50 million people are expected to reach menopause each year.
Whilst a natural part of ageing, menopause often has a significant impact on personal, social and professional quality of life. Alarmingly, research from the Fawcett Society indicates that 10% of women have left the workplace due to the symptoms of menopause whilst a 2019 survey found that three in five menopausal people were negatively affected at work.
Classic symptoms of menopause include hot flushes (vasomotor symptoms), mood changes such as anxiety and depressive symptoms, urogenital symptoms including sexual dysfunction, incontinence and urinary tract infections, disturbed sleep, impaired verbal memory and musculoskeletal pain. For many, these symptoms are severe and debilitating. Even those who do not experience severe symptoms are at risk of long-term effects including increased risk of cardiometabolic disease, diabetes, bone loss and cancers associated with increased abdominal fat (e.g., breast, colon and endometrial cancer).
Symptoms are likely underdiagnosed and undertreated. For many, hot flushes are the only symptom they attribute to menopause, meaning that other symptoms are left inadequately managed. Many are hesitant to ask for help in case their symptoms are dismissed by medical professionals as a normal part of ageing.
Nevertheless, pharmacological treatments are available. The gold standard treatment for menopause symptoms is hormone replacement therapy (HRT), which aims to replace hormones lost during the menopause transition (i.e., oestrogen and progesterone). However, patient confidence in the safety of HRT was severely damaged following the publication of the 2002 Women’s Health Initiative (WHI) study and the 2003 UK Million Women Study (MWS) which first linked HRT to increased risk of blood clots and stroke and a small absolute increased risk of breast cancer respectively. The resultant decline in HRT prescribing cast doubt over the commercial returns within the women’s health sector. As such, pharmacological innovation for menopause treatment has largely been limited to novel combinations of oestrogen and progesterone (e.g. synthetic or bioidentical forms) and formulations optimised for different routes of administration (e.g., oral tablets, patches, suppositories, gels or sprays).
More recently, interest in the menopause market has been piqued by the success of novel nonhormonal therapies such as fezolinetant. This next generation of therapeutics stems from the research of neuropathologist Prof Naomi Rance, who identified a specific group of neurons in the hypothalamus (a part of the brain that controls body temperature) that become overactive and enlarged in the brains of menopausal people. However, whilst fezolinetant has been approved by the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) and is currently available via private prescription, it is not yet available via the National Health Service (NHS).
Faced with “medical gaslighting” and a dearth of suitable therapeutics, tech-savvy women are increasingly taking matters into their own hands and turning to technology to ease their menopause symptoms; the result being a boom in the FemTech (“female technology”) industry and rich pickings for innovators.
A notable example is Vira Health’s Stella app, which aims to provide a personalised treatment plan (for example, cognitive behavioural therapy, sleep scheduling, pelvic floor exercises) based on specific symptoms, machine learning, and lifestyle preferences. Devices aimed at relieving the symptoms of hot flushes include the Menopod, which was featured on an episode of Canadian Dragons Den, and the Thermaband Zone.
In such a rapidly growing market, innovators would be wise to develop a strategy for protecting and exploiting their intellectual property at an early stage. If in doubt, your patent attorney will be able to guide you.
Mathys & Squire have ranked in the 2025 Chambers and Partners UK Legal Guide, which acknowledges some of the top ranked law firms and lawyers each year. The research analysts obtain information through one-to-one interviews and investigation.
We are delighted to announce that Mathys & Squire have maintained our ranking in the Chambers and Partners 2025 guide.
Chambers state:
“The Mathys & Squire team are able to handle complex and sophisticated matters with extreme competence and professionalism in a timely manner.”
“Mathys & Squire are always responsive and apply very high standards to get to the final product. They are nothing short of outstanding.”
“Mathys & Squire combines good technical patent work with pragmatism and successfully leverages technical and industry knowledge.”
Amongst the team, Partner Martin Maclean has also been ranked as a Band 1 Patent Attorney, acknowledged for his work as an “outstanding patent attorney.”
For more information on the Mathys & Squire rankings, visit the Chambers and Partners website here.
In a much-anticipated decision, Technical Board of Appeal 3.3.04 has held that there is no requirement under the EPC for an applicant to amend the description of a patent application before grant (T 56/21). This case is the latest in a developing line of decisions from the EPO’s Technical Boards of Appeal to go against the Office’s long-standing practice of requiring amendments to the description before grant for consistency with the claims.
Of particular interest, the Board declines to refer questions to the Enlarged Board of Appeal which could help resolve this issue, despite having indicated its intention to do so earlier in appeal proceedings. In a detailed decision (running to 90 pages), the Board concludes that a referral is unnecessary, among other things because of recent developments from the Court of Appeal of the UPC and because it views the EPO case law as “evolving” rather than divergent. Ultimately, the Technical Board takes the view that “[t]he wording of Article 84 EPC as well as its context leave no room for requiring, in examination, that the description be adapted to allowable claims to match their subject-matter.” The Board takes the clear view that Article 84 is a requirement to be met by the claims and cannot impose any conditions on the content of the description. Rules 42, 43 and 48, which have previously been suggested by some as a legal basis for requiring description amendments, are also dismissed.
A notable feature of the decision is that the Board sees a clear separation between the assessment of claims for compliance with the clarity and support requirements under Article 84 EPC on the one hand, and on the other hand the principles governing the interpretation of those claims post-grant under Article 69 EPC to determine of the extent of protection which they confer. The Board concludes that Article 69 EPC has no relevance in grant proceedings, and criticises the EPO’s current practice as “encroach[ing] on the competence of national courts and legislators” in seeking to use description amendments to “reduce the variability in the determination of the extent of protection of a patent … and to arrive at a more “harmonised” determination of the extent of protection”. Given that the relevance of Article 69 is already the subject of a pending referral to the Enlarged Board (G 1/24), it seems surprising that the Board in this instance has reached such an unequivocal conclusion. This suggests that the Board in the present instance might be seeking to influence that debate, and raises a question as to whether its view on description amendments might change if the Enlarged Board does not see such a clear separation between the rules for claim interpretation pre- and post-grant.
This decision provides applicants with further legal support for refusing an order to amend a description before grant, but it seems unlikely that the EPO will change its Guidelines or its practice unless and until a referral to the Enlarged Board is eventually made on this topic.
In a surprise move, the representatives acting on behalf of UC Berkley defending CRISPR patents in appeal proceedings before the EPO have withdrawn their approval of the texts of the patents thereby revoking them and bringing the appeal proceedings to an end. When doing so, the representatives justified the withdrawal referencing “serious procedural concerns” arising from the Board of Appeal handling of appeal T2229/19, a decision Mathys & Squire are seeking to reverse through an application for Petition for Review.
In T2229/19 a Board held as inadmissible a request to delete two dependent claims solely on the grounds that although the deletion would have addressed all the objections considered so far, the deletion would not prima facie overcome the Board’s preliminary opinion on a different, yet to be discussed issue, namely sufficiency of disclosure.
In the CRISPR appeal, UC Berkely’s representatives concluded that given that the chairperson and rapporteur in their appeal were also members of the Board in T2229/19, the risk that the Board would adopt a similar approach in their appeal was such that the Patentees could not “be expected to expose the Nobel-prize winning invention protected by the … patent[s] to the repercussions of a decision handed down under such circumstances, when other members [of the patent family were] still at a stage where the Patentees [could] ensure that they [would] ultimately be fully heard on all substantive issues.”
It is highly unusual for a patentee to withdraw their approval of the text of a patent during appeal proceedings and accompany such a withdrawal with an expansive statement as to why a patent is valid and why the patentees have concerns about the expected handling of a case by a Board of Appeal.
It could well be that UC Berkley were merely providing cover for a withdrawal which will significantly delay the EPO making a final ruling on the validity of a patent. Certainly, that was the view of the representatives of the opponents who described the withdrawal as “a transparent move to avoid any possibility of an adverse decision” which they claimed was “justified by a (spurious and hypothetical) concern about the future conduct of the Board.”
However, if UC Berkley’s concerns are to be taken at face value, they raise serious questions about the EPO Boards of Appeal and in particular the Rules of Procedure which were introduced in 2020.
Those Rules of Procedure sought to emphasise that EPO Appeals should be restricted to a review of the proceedings at first instance and reduce the extent to which new matters might be raised on appeal. However, it is evident, that in some areas this has led to inconsistencies in approach. In our Petition for Review we noted that two distinct approaches [1] to the deletion of dependent claims during appeal proceedings (neither of which had been followed by the Board in that case) had arisen.
UC Berkely noted that our Petition for Review against T2229/19 could be pending for quite some time. Although the Petition for Review procedure was intended to be as simple and short as possible and prior to 2020, Petitions for Review were typically resolved in around 10 months, evidently that is no longer the case as was clearly demonstrated with the four-and-a-half year time period taken to resolve the Petition for Review in R1/20 which was finally resolved in July this year.
In the meantime, even though a Petition for Review is pending against it, T2229/19 serves as a potential precedent to be followed in other EPO Appeals and indeed recently another EPO Board of Appeal in T172/22 referred to T2229/19 as a precedent for refusing to allow claims to be deleted during appeal proceedings.
When Board of Appeal decisions result in the filing of Petitions for Review and cause the withdrawal of high-profile appeals, the apparent inability of the EPO to rule on such Petitions in a timely manner is very concerning.
[1] T1480/16 vs T1224/15
We are delighted to announce that Mathys & Squire has upheld its position in the 2025 edition of The Legal 500 for both PATMA: Patent Attorneys and PATMA: Trade Mark Attorneys categories.
Patent Partners Chris Hamer, Alan MacDougall, Martin MacLean, Jane Clark, Paul Cozens, Dani Kramer, Philippa Griffin, James Wilding, Sean Leach, James Pitchford, as well as Alexander Robinson, Peter Arch, Laura Clews, who are newly ranked, are all featured in the 2025 edition of the directory.
Mathys & Squire’s trade mark team has also received recognition in the directory. From our trade mark practice, Partners Margaret Arnott, Rebecca Tew andGary Johnston, andHelen Cawley who is listed, and Managing Associate Harry Rowe, have been ranked.
The firm received glowing testimonials for its patent and trade mark practices:
”M&S have a very deep technical and legal experience that covers areas very relevant for green carbon sequestration technologies. They also have representation in key markets in Germany and the UK, which makes it very relevant. The team work very well together and drafted in a quick time period our patent application.”
”M&S team of IP experts provides unparalleled knowledge and experience in all areas of intellectual property, including patents, trademarks, design protection, and litigation. The firm’s versatile team of attorneys, scientists, and strategists possesses deep experience navigating complex IP landscapes for high-growth, technology-driven industries.”
”The Mathys & Squire team have been excellent in providing focused and pertinent advice on tricky issues and have shown considerable technical and commercial ability in fintech patent issues.”
”From my first call till today, I have seen a kind and professional approach from Mathys & Squire. Despite my language barriers, their patience and understanding makes me feel comfortable and confident through the communication process. I have worked on two patent applications with them in the last year.”
“The entire team at Mathys & Squire is very professional, welcoming and very patient. We pose some difficult issues sometimes and we always get a prompt, detailed response which enables us to take a final decision with the client. We are very pleased both professionally and administratively.”
“I’ve worked with Mathys & Squire for the last 15 years. They mostly deal with our patent work and some design registration. I’m impressed with the way they deal with all our patent office issues. On a personal level, they are easy to work with and, when we write new patents, they make the process straightforward and painless.”
“I have not found any scientific area of focus where [the] team aren’t comfortable. The Mathys & Squire team do excellent legal work for us and are always on the lookout for information that will help further our understanding of European legal issues.”
”Paul Cozens demonstrates a masterful grasp of the subject area and is solution driven to problem solving.”
”Alan MacDougall has been my trusted business partner for more than 10 years. Alan is diligent person who provides helpful, accurate, and timely answers to questions that are unclear for us. Alan has a wealth of knowledge and experience in the intellectual property field and his useful and appropriate advices has always been of great help to us. We look forward to working with him to further expand our business.”
“If intellectual property is or could be important to your business, and you want to be assured of the best possible advice, then look no further than Mathys & Squire and Dani Kramer in particular, you will be hard pressed to find any better.”
”Sean Leach is technically and legally competent, approachable and responsive.”
“The trade mark team is supportive, available, and interested in supporting the business to grow.”
”Gary Johnston has oodles of experience and is sensible and commercial.’
”Harry Rowe is fantastic and really helps us to secure trade marks and to pursue other IP protections. Harry has integrity and is genuinely interested in our business, which is a tech-start with ambition for global growth and societal change. He gets the importance of the latter also. He helps us to map a pathway forward to support our business to grow.”
”Harry Rowe has excellent communication skills, often having to translate complicated legal agreements for me in a way that I will understand them. He makes sure to get a very clear understanding of what’s unique about our business and can think laterally to identify issues that might become an issue for us in the future.”
For full details of our rankings in The Legal 500 2025 guide, please click here.
We extend our gratitude to all our clients and connections who participated in the research, and we extend our congratulations to our individual attorneys who have earned rankings in this year’s guide.
Managing IP have released their guide for the 2024 IP Rising Stars, which recognises the best up-and-coming practitioners across several IP areas and more than 50 jurisdictions. Each year, the research analysts obtain information through firm submissions, research data and feedback to identify the IP Rising Stars.
We are delighted to announce that Partners Edd Cavanna and Max Thoma and Managing Associate Harry Rowe have been named as 2024 IP Rising Stars.
Alongside IP STARS, Managing IP annually identifies professionals who have demonstrated excellent practice on behalf of their firm, making it a commendable award.
For more information on the Mathys & Squire rankings, visit the IP Stars website here.